Introducción
El sistema del complemento es uno de los principales componentes de la inmunidad innata. Su activación se produce a través de tres vías: la clásica, dependiente de inmunoglobulinas unidas a C1q; la de lectinas, mediada por la lectina fijadora de manosa; y la alternativa, que además de actuar de forma independiente potencia a las otras vías. Todas convergen en la formación de convertasas de C3 y C5, encargadas de la producción de fragmentos con diferentes funciones: C3a y C5a actúan como mediadores proinflamatorios, C3b como opsonina y amplificador de la cascada, y el proceso culmina con el complejo de ataque a membrana (C5b-9), el cual es capaz de inducir lisis celular1–3. Este sistema cuenta con diferentes mecanismos de regulación, tanto solubles como de superficie, incluidos el factor H, el factor I y las proteínas CD46, CD55 y CD59, que limitan la activación y protegen los tejidos. Las alteraciones en estos reguladores y autoanticuerpos, como el factor nefrítico C3 (C3NeF), favorecen la activación descontrolada que ocasiona inflamación y daño renal1–3. La glomerulopatía por C3 (GC3) representa un espectro de enfermedades renales raras caracterizadas por depósitos dominantes de C3 en la inmunofluorescencia4–6. Incluye tres afecciones principales: 1) glomerulonefritis por C3 (GNC3), con depósitos mesangiales y subendoteliales; 2) enfermedad por depósitos densos (EDD), definida por depósitos intramembranosos hiperdensos; y 3) nefropatía asociada a CFHR5, descrita en familias chipriotas con curso hereditario. En cuanto a sus manifestaciones clínicas, la GC3 puede presentarse a cualquier edad, la EDD es más común en la infancia y la adolescencia, y la GNC3 predomina en adultos jóvenes y de mediana edad6–9. Su expresión clínica es heterogénea e incluye proteinuria en rangos variables (desde subnefrótica hasta síndrome nefrótico), hematuria persistente o macroscópica intermitente, hipertensión arterial y, en algunos casos, lesión renal aguda o crónica con elevación inicial de la creatinina4,6,9.
Caso clínico
Varón de 29 años que acudió a consulta en septiembre de 2020 por orina espumosa y elevación intermitente de la presión arterial. No se conocían antecedentes familiares relevantes, ya que el paciente era adoptado. En la exploración física inicial, como único dato relevante destaca una presión arterial de 130/90 mmHg. Los estudios de laboratorio mostraron parámetros hematológicos normales; creatinina 1.4 mg/dl, urea 58 mg/dl, colesterol total 240 mg/dl, triglicéridos 190 mg/dl, albúmina 3.9 g/dl y ácido úrico 9 mg/dl. La proteinuria en orina de 24 horas fue de 780 mg y la albuminuria de 230 mg. El examen general de orina reportó densidad 1.015 y pH 6.5, sin hematuria ni cilindros.
Por la edad del paciente y la presencia de proteinuria, inicialmente se sospechó una glomeruloesclerosis focal y segmentaria, por lo que se realizó una biopsia renal percutánea en octubre de 2020. El reporte mostró un patrón de glomerulonefritis proliferativa mesangial difusa y esclerosis parahiliar focal (Fig. 1). En la inmunofluorescencia se observó C3c (2+) mesangial, con IgA débil (1+) y negatividad para IgG, IgM, C1q, fibrinógeno y cadenas ligeras. El nivel sérico de C3 fue de 110 mg/dl. No se contó con microscopía electrónica, lo que constituye una limitación al impedir la diferenciación formal entre los subtipos de la patología; en consecuencia, el caso se reporta como GC3. No se realizaron estudios ampliados de complemento ni análisis genéticos (C3NeF, anti-FH, secuenciación de genes reguladores del complemento).
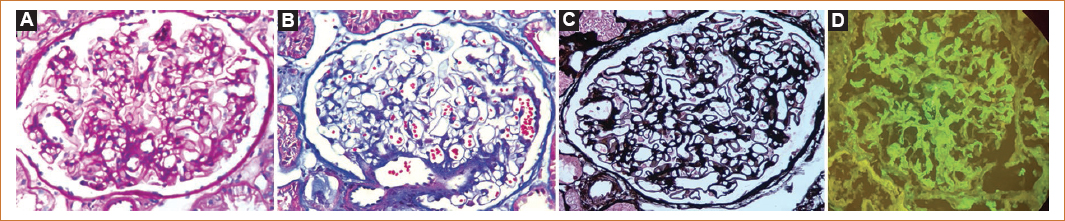
Figura 1. Biopsia renal. Tinciones de ácido periódico de Schiff (A), tricrómico de Masson (B) y metenamina de Jones (C), 40×. En las tres se observa un ovillo glomerular con luces capilares abiertas e hipercelularidad mesangial leve (≤ 5 células por espacio mesangial), sin alteraciones evidentes de la membrana basal. La inmunofluorescencia (D) para C3c muestra depósitos granulares mesangiales y subendoteliales.
Se inició tratamiento con pravastatina 10 mg/día, losartán 6.25 mg/día y alopurinol 150 mg tres veces por semana, y un plan nutricional hiposódico, normoproteico (0.8 g/kg/día, con predominio de proteínas vegetales), restricción de fósforo y control de líquidos. Durante el seguimiento se observó respuesta favorable, con mejoría de la proteinuria y preservación de la función renal. La evolución bioquímica mostró valores estables a lo largo de 5 años (2020-2025), con creatinina 0.9-1.2 mg/dl, urea 38-50 mg/dl, proteinuria en rango subnefrótico y niveles de C3 sérico dentro de la normalidad. En todo el seguimiento el paciente se mantuvo asintomático, sin episodios de hematuria, edema, deterioro progresivo de la función renal ni complicaciones extrarrenales (Tabla 1). En enero de 2024 se añadió dapagliflozina 10 mg/día y en agosto del mismo año se cambió el tratamiento de hiperuricemia a febuxostat 80 mg/día. El paciente no desarrolló complicaciones extrarrenales ni deterioro progresivo de la función renal. Hasta enero de 2025, tras más de 5 años de seguimiento, el paciente permanece con función renal estable, proteinuria subnefrótica y control metabólico adecuado bajo manejo conservador, sin necesidad de inmunosupresión ni terapias dirigidas al complemento. Se observa estabilidad en la creatinina sérica y proteinuria en rango subnefrótico durante todo el seguimiento, con niveles de C3 sérico dentro de la normalidad.
Tabla 1. Evolución de los parámetros bioquímicos y de la proteinuria durante el seguimiento (2021-2025)
| Fecha | Urea (mg/dl) | Creatinina (mg/dl) | Colesterol (mg/dl) | Ácido úrico (mg/dl) | Albúmina (g/dl) | Proteinuria (mg/24h) | Albuminuria (mg/24h) | C3 (mg/dl) |
|---|---|---|---|---|---|---|---|---|
| Enero 2021 | 50 | 1.2 | 190 | 6.0 | 4.1 | 320 | 110 | 101 |
| Agosto 2021 | 48 | 1.1 | 180 | 7.0 | 4.0 | 280 | 140 | 130 |
| Enero 2022 | 42 | 1.0 | 200 | 6.4 | 4.3 | 210 | 130 | 140 |
| Agosto 2022 | 40 | 1.0 | 210 | 5.8 | 4.4 | 290 | 135 | 135 |
| Enero 2023 | 38 | 0.9 | 180 | 5.6 | 4.2 | 220 | 115 | 156 |
| Agosto 2023 | 43 | 1.0 | 190 | 6.0 | 3.6 | 315 | 125 | 160 |
| Enero 2024 | 45 | 1.1 | 210 | 6.9 | 3.7 | 340 | 158 | 105 |
| Agosto 2024 | 46 | 1.0 | 250 | 8.5 | 4.6 | 290 | 150 | 94 |
| Enero 2025 | 45 | 1.1 | 200 | 6.8 | 4.0 | 350 | 148 | 90 |
Discusión
La GC3 se caracteriza por un curso clínico variable. En la mayoría de las cohortes, el 30-50% de los pacientes progresan a enfermedad renal crónica terminal en la primera década tras el diagnóstico, con recurrencia de alrededor del 90% después del trasplante y pérdida temprana del injerto en el 30-70% de los casos6,9,10. Aunque la EDD suele asociarse a un curso más agresivo y la GNC3 a una evolución más lenta, ambas comparten un pronóstico globalmente desfavorable4,8. Resulta relevante, por lo tanto, documentar trayectorias atípicas, ya que permiten resaltar la heterogeneidad de la enfermedad y orientar hacia la identificación de factores pronósticos y a la selección terapéutica adecuada. El caso descrito, con estabilidad clínica y funcional por más de 5 años, ejemplifica esta variabilidad.
El diagnóstico se establece mediante biopsia renal; en la microscopía óptica, el hallazgo más común corresponde a un patrón membranoproliferativo, caracterizado por proliferación mesangial y endocapilar, presencia de doble contorno y lobulación, y en algunos casos puede observarse un patrón esclerosante, descrito con mayor frecuencia en la GNC37–9. La inmunofluorescencia confirma la dominancia de C3, mientras que la microscopía electrónica permite clasificarla adecuadamente según las características antes mencionadas4,6–9. Los estudios complementarios incluyen pruebas serológicas, siendo la hipocomplementemia con C3 bajo y C4 normal un hallazgo característico; sin embargo, hasta el 40% de los pacientes pueden mostrar valores normales, como se apreció en la serología reportada. El diagnóstico diferencial se hará con la glomerulonefritis posinfecciosa, ya que ambas pueden presentar depósitos dominantes de C3 en la inmunofluorescencia. A diferencia de la GC3, la posinfecciosa se suele asociar a inmunocomplejos y en general muestra resolución clínica y normalización de los niveles de complemento en el transcurso de pocas semanas7,8,10.
Los factores de mal pronóstico son la edad avanzada, el síndrome nefrótico, la hipertensión persistente, la proteinuria mantenida, el filtrado glomerular bajo al inicio y las lesiones crónicas en la biopsia (esclerosis y fibrosis)4,6,8,9. La evaluación integral puede plantearse en dos dimensiones: estratificación basal y dinámica longitudinal. En la basal, un estudio multicéntrico desarrolló un nomograma con tres variables sistemáticas (tasa de filtración glomerular estimada [TFGe], proteinuria y cronicidad histológica), que permite estimar la supervivencia renal hasta 10 años11. Así, una TFGe conservada, una proteinuria baja y una mínima cronicidad se asocian a mejor evolución. Nuestro paciente, con TFGe estable (creatinina 0.9-1.2 mg/dl), proteinuria subnefrótica y asintomático, encaja en este perfil. La biopsia mostró esclerosis parahiliar focal, asociada a peor evolución en cohortes recientes12,13. Sin embargo, el peso mayor de la proteinuria baja y la función renal preservada parece haber predominado en este caso. Un análisis demostró que cada aumento de 1 g/dl de proteinuria eleva el riesgo de fallo renal, mientras que reducciones ≥ 30% a 6 meses y ≥ 50% a 12 meses se asocian a mejor supervivencia. Mantener la proteinuria < 1 g/dl promedio predice una evolución favorable. El perfil descrito, con proteinuria persistentemente subnefrótica y TFGe estable, se alinea con los parámetros de bajo riesgo reportados13.
El tratamiento continúa siendo un desafío. Las medidas generales incluyen el control estricto de la presión arterial y el bloqueo del sistema renina-angiotensina-aldosterona, los cuales se asocian a mejores desenlaces. El micofenolato de mofetilo combinado con glucocorticoides es el esquema inmunosupresor más utilizado, aunque con eficacia variable, mientras que la plasmaféresis ha mostrado resultados inconsistentes5,6,8–10. El eculizumab, un anticuerpo anti-C5, se ha empleado en formas rápidamente progresivas o con actividad terminal elevada (sC5b-9), con beneficios modestos y recaídas frecuentes10,14. Hasta la fecha no existe un inhibidor del complemento aprobado específicamente para la GC3; sin embargo, nuevos agentes proximales han mostrado resultados alentadores. Por ejemplo, el pegcetacoplán redujo aproximadamente un 50% la proteinuria y elevó el C3 sérico en un ensayo de fase 2. Estos hallazgos refuerzan el potencial de la inhibición del complemento, aunque persisten retos para establecer un estándar terapéutico, dadas la rareza y la heterogeneidad de la enfermedad y la dificultad de realizar ensayos amplios12. Un componente emergente del soporte son los inhibidores del cotransportador de sodio y glucosa tipo 2 (SGLT2); aunque la evidencia específica en GC3 es limitada, los ensayos DAPA-CKD y EMPA-KIDNEY demostraron nefroprotección en enfermedad renal crónica no diabética, incluidas glomerulonefritis, con reducciones de la proteinuria y un buen perfil de seguridad, y estudios de cohorte han mostrado su beneficio en pacientes no diabéticos15. La introducción de la dapagliflozina en nuestro escenario refuerza la estabilidad clínica alcanzada bajo manejo conservador.
Limitaciones
Como antes hemos mencionado, no se contó con microscopía electrónica ni con estudios complementarios de complemento y genética (C3NeF, anti-FH, variantes en CFH/CFI/CFB/C3/CFHR), los cuales son recomendados por los consensos recientes. Esta limitación impide establecer con certeza la distinción entre EDD, GNC3 u otras variantes, y restringe la posibilidad de una adecuada estratificación biológica. No obstante, la estabilidad clínica y analítica a lo largo del seguimiento respalda el enfoque conservador adoptado en nuestro paciente.
Conclusión
La heterogeneidad y la incertidumbre pronóstica de la GC3 obligan a mantener una vigilancia estrecha y un enfoque terapéutico individualizado. Recomendamos estratificar la TFGe, la proteinuria y la cronicidad histológica para anticipar el riesgo y guiar la intensidad terapéutica, dar seguimiento a la proteinuria como marcador útil de eficacia y pronóstico, y manejo integral basado en bloqueo del sistema renina-angiotensina-aldosterona, control metabólico e inhibidores de SGLT2, y reservar el uso de terapias específicas dirigidas al complemento de manera individualizada.
Financiamiento
Los autores declaran que este trabajo se realizó con recursos propios.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Consideraciones éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad, consentimiento informado y aprobación ética. Los autores han obtenido la aprobación del Comité de Ética para el análisis de datos clínicos obtenidos de forma rutinaria y anonimizados. Debido a la naturaleza del estudio, no fue necesario el consentimiento informado individual. Se han seguido las recomendaciones éticas pertinentes.
Declaración sobre el uso de inteligencia artificial (IA). Los autores declaran que no se utilizó ningún tipo de inteligencia artificial generativa para la redacción ni la creación de contenido de este manuscrito.
